
Case report- a case of Creutzfeldt-Jakob disease
Abstract
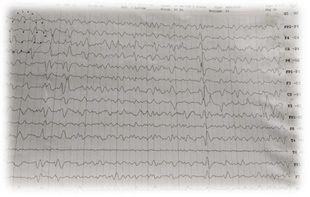
Creutzfeldt-Jakob Disease (CJD) is a rare invariably fatal neurodegenerative disease believed to be caused by an abnormal isoform of cellular infectious glycoprotein called prion protein. Though it is arare disease; yet it is the most common among prion diseases. Clinical presentation consists of rapidly progressive loss of memory, cognitive & visual disturbance, lack of coordination, myoclonus, cerebellar, pyramidal and extra pyramidal signs, akineticmutism & with progression of disease deterioration in higher mental functions become more pronounced. Periodic sharp triphasic wave complexes on EEG, high signal abnormalities in caudate nucleus and putamen on diffusion weighted (DW) or FLAIR MRI of Brain and positive 14-3-3 protein in CSF substantiate the diagnosis of CJD but definitive diagnosis is established by brain biopsy or autopsy materials. We report a case of 58-year old female patient who was admitted with complaints of rapidly progressive dementia, cognitive disturbance, blurring of vision and myoclonic jerks. Initial MRI brain and CSF findings were normal. Differential diagnoses that can present with rapidly progressive dementia and thereby mimic sporadic Creutzfeldt-Jakob disease were considered after review of literature. In EEG triphasic wave complexes were seen, repeat DWMRI after two weeks showed bilateral hyper-intensities in basal ganglia involving caudate nucleus and putamen, suggesting a diagnosis of probable CJD on the basis of center for disease control and prevention (CDC) criteria. The case is reported because of its rarity and also to emphasise that patients with rapidly progressive dementia, associated visual and cognitive disturbances and myoclonus should be investigated with DW MRI, EEG&CSF for diagnosis of CJD.
Downloads
References
Chen C, Dong XP. Epidemiological characteristics of human prion diseases. Infect Dis Poverty. 2016;5(1):47. DOI: https://doi.org/10.1186/s40249-016-0143-8.
Rabinovici GD, Wang PN, Levin J, Cook L, Pravdin M, Davis J, First symptom in sporadic Creutzfeldt-Jakob disease. Neurol. 2006;66(2):286-7. DOI: https://doi.org/10.1212/01.wnl.0000196440.00297.67
Young GS, Gesch wind MD, Fischbein NJ, Martindale JL, Henry RG, Liu S, et al. Diffusion-weighted and fluid-attenuated inversion recovery imaging in Creutzfeldt-Jakob disease: high sensitivity and specificity for diagnosis. AJNR Am J Neuroradiol. 2005; 26(6):1551-62.
Hill AF, Joiner S, Wadsworth JD, Sidle KC, Bell JE, Budka H, et al. Molecular classification of sporadic Creutzfeldt-Jakob disease. Brain. 2003;126 (Pt 6):1333-46. DOI: https://doi.org/10.1093/brain/awg125
Krasnianski A, Bohling GT, Harden M, Zerr I. Psychiatric symptoms in patients with sporadic Creutzfeldt- Jakob disease in Germany. J Clin Psychiatry. 2015; 76(9):1209-15. DOI: https://doi.org/10.4088/jcp.13m08915.
Collins SJ, Sanchez-JuanP, Masters CL, Klug GM, van Duijn C, Poleggi A, et al. Determinants of diagnostic investigation sensitivities across the clinical spectrum of sporadic Creutzfeldt-Jakob disease. Brain. 2006; 129(Pt 9):2278-87. Epub 2006 Jul 1. DOI: https://doi.org/10.1093/brain/awl159
Nakatani E, Kanatani Y, Kaneda H, Nagai Y, Teramukai S, Nishimura T, et al. Specific clinical signs and symptoms are predictive of clinical course in sporadic Creutzfeldt-Jakob disease. Eur J Neurol. 2016; 23 (9):1455-62. DOI: https://doi.org/10.1111/ene.13057. Epub 2016 May 24.
Mehndiratta MM, Bajaj BK, Gupta M, Anand R, Tatke M, et al. Creutzfeldt-Jakob disease: Report of 10 cases from North India. Neurol India. 2001; 49(4):338-41.
Velásquez-Pérez L, Rembao-Bojorquez D, Guevara J, Guadarrama-Torres RM, Trejo- Contreras A. Creutz feldt - Jakob disease in Mexico. Neuropathol. 2007; 27 (5):419-28. DOI: https://doi.org/10.1111/j.1440-1789.2007.00807.x
González-Duarte A, Medina Z, Balaguer RR, Calleja JH. Can prion disease suspicion be supported earlier? Clinical, radiological and laboratory findings in a series of cases. Prion. 2011;5(3):201-7. DOI: https://doi.org/10.4161/pri.5.3.16187. Epub 2011 Jul 1.
Chandra SR, Issac TG, Philip M, Gadad V. Creutz feldt-Jakob Disease Phenotype and Course: Our Experience from a Tertiary Center. Indian J Psychol Med. 2016; 38 (5): 438-442. DOI: https://dx.doi.org/10.4103%2F0253-7176.191376
Mahale RR, Javali M, Mehta A, Sharma S, Acharya P, Srinivasa R. A study of clinical profile, radiological and electroencephalographic characteristics of suspected Creutzfeldt-Jakob disease in a tertiary care centre in South India. J Neurosci Rural Pract. 2015; 6(1): 39-50. DOI: https://dx.doi.org/10.4103%2F0976-3147.143189.
Biswas A, Khandelwal N, Pandit A, Roy A, Guin DS, Gangopadhyay G, et al. Case series of probable sporadic Creutzfeldt-Jakob disease from Eastern India. Ann Indian Acad Neurol. 2013;16(4):659-63. DOI: https://dx.doi.org/10.4103%2F0972-2327.120486.
Pasha SA, Ankamma Rao, Phani Bhushan, Bhaskara Rao B, Nayak P (2017) Case Report of Clinically Probable Sporadic Creutzfeldt - Jakob Disease from A Tertiary Care Hospital in South India. Ann Alzheimers Dement Care 2(1): 013-017.DOI: https://doi.org/10.17352/aadc.000003.
Zerr I, Pocchiari M, Collins S, Brandel JP, de Pedro Cuesta J, Knight RS, et al. Analysis of EEG and CSF 14-3-3 proteins as aids to the diagnosis of Creutzfeldt-Jakob disease. Neurol. 2000; 55(6):811-5. DOI: https://doi.org/10.1212/WNL.55.6.811
Muayqil T, Gronseth G, Camicioli R. Evidence-based guideline: diagnostic accuracy of CSF 14-3-3 protein in sporadic Creutzfeldt-Jakob disease: report of the guideline development subcommittee of the American Academy of Neurology. Neurol. 2012;79 (14):1499-506. DOI: https://doi.org/10.1212/WNL.0b013e31826d5fc3. Epub 2012 Sep 19.
Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heinemann U, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain. 2009;132(Pt 10):2659-68. DOI: https://doi.org/10.1093/brain/awp191. Epub 2009
Green AJE. RT-QuIC: a new test for sporadic CJD. Pract Neurol. 2019; 19(1): 49-55. DOI: http://dx.doi.org/10.1136/practneurol-2018-001935. Epub 2018
Brown P, Gibbs CJ Jr, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, et al. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994; 35(5): 513-29. DOI: https://doi.org/10.1002/ana.410350504.

 OAI - Open Archives Initiative
OAI - Open Archives Initiative