A Case Series Report on Congenital pulmonary airway malformation – antenatal diagnosis and post-natal follow up
Sahu S1, Khokan M2, Guha Roy A3*
DOI:https://doi.org/10.17511/ijmrr .2024.i04.06
1 Suparna Sahu, Senior Resident, Department of Radiodiagnosis, Nil Ratan Sircar Medical College, Kolkata, West Bengal, India.
2 Mandi Khokan, Demonstrator, Department of Radiology, Deben Mahato Government Medical College and Hospital, Purulia, West Bengal, India.
3* Anik Guha Roy, Post Graduate Resident, Md Radiodiagnosis, Nil Ratan Sircar Medical College, Wbuhs, West Bengal, India.
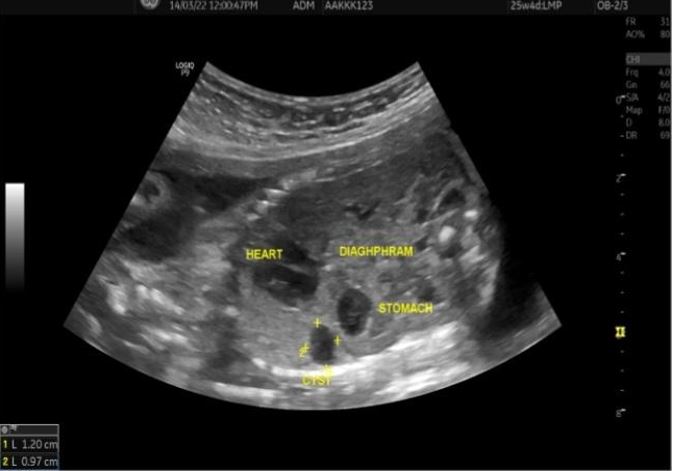
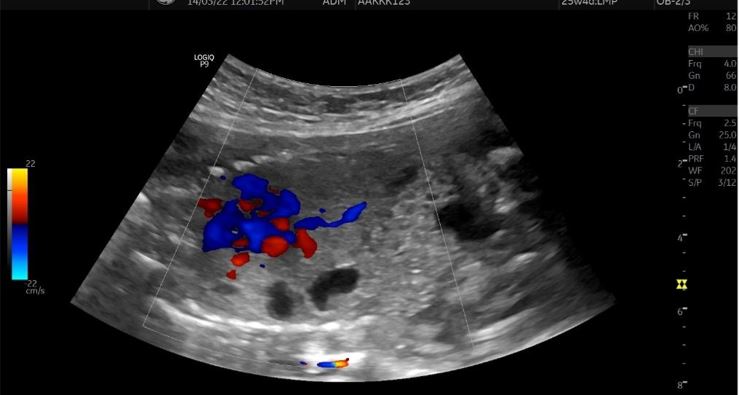
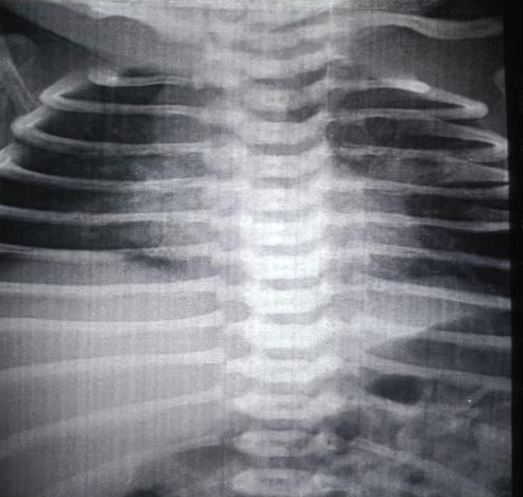
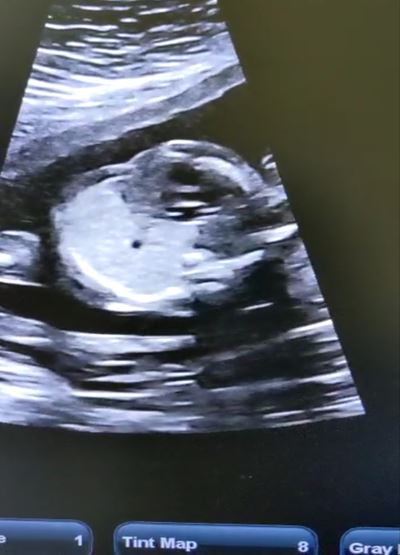
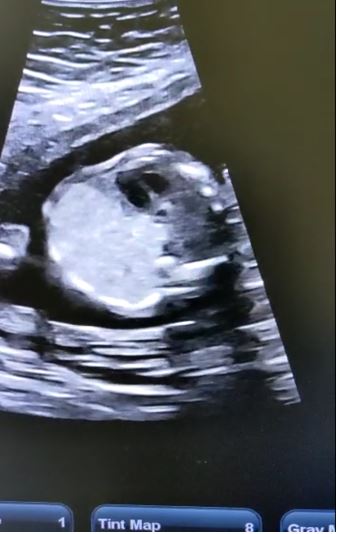
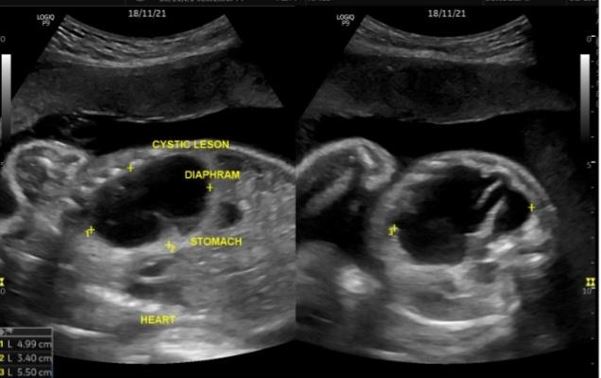
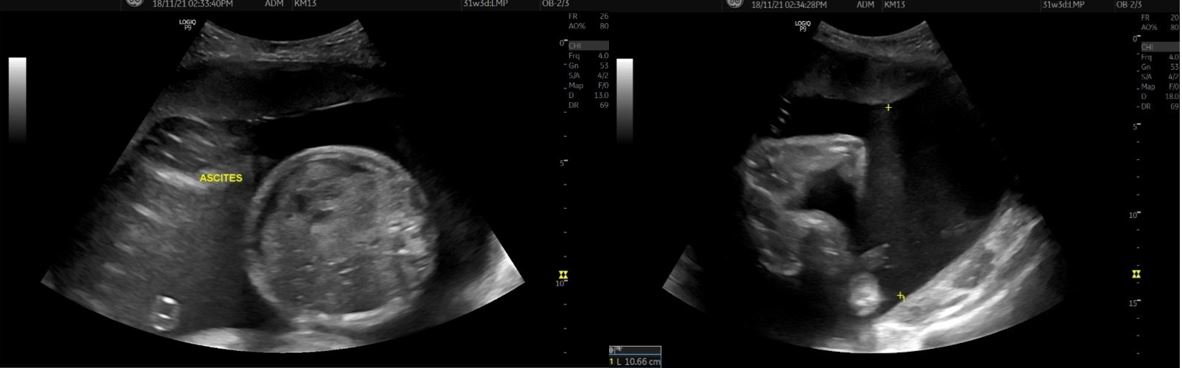
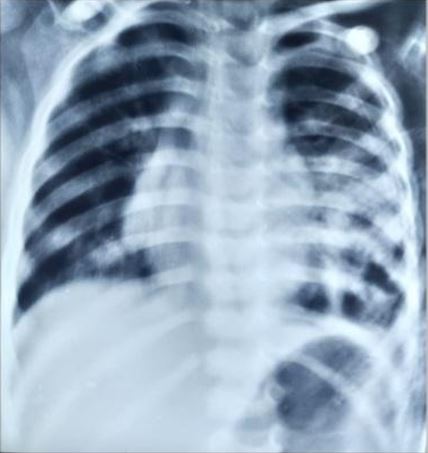
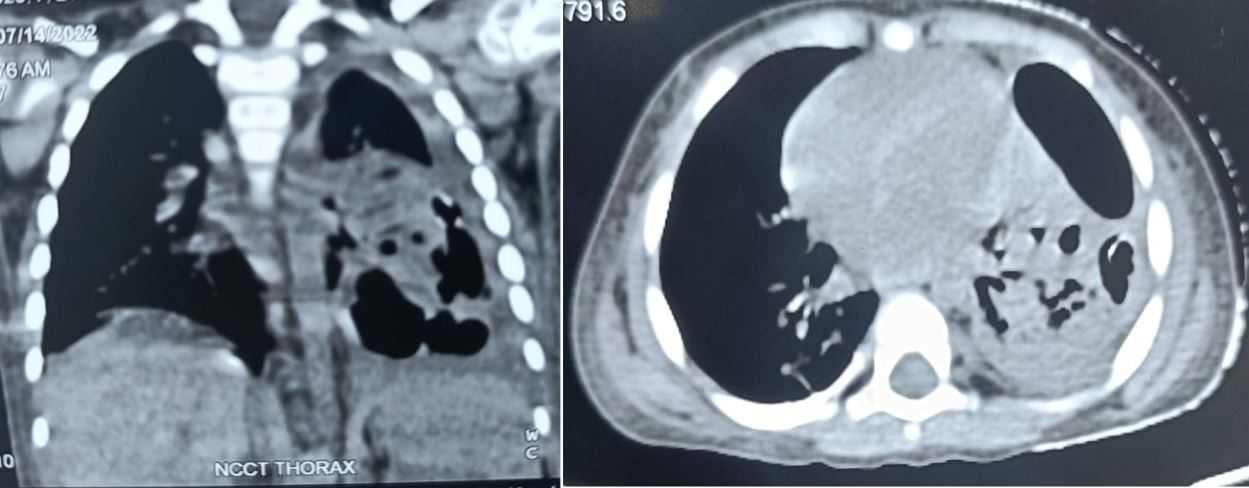
Congenital pulmonary airway malformation is the most commonly detected congenital lung defect in the ante-natal period. CPAM is a rare developmental, non-hereditary dysplastic lung lesion. No obvious association with maternal age, race or exposure to any given factor could be ascertained. It has an incidence of about 1:1500-4000 of all live births and accounts for approximately 25% of all congenital lung lesions. Abnormal proliferation of the fetal tracheobronchial tree in the early weeks of gestation can lead to the development of this condition. Frequent associated congenital defects include congenital heart defects, tracheoesophageal fistula, congenital diaphragmatic hernia, and renal agenesis. Post-natal prognosis of CPAM depends on the size of the lesion, degree of pulmonary hypoplasia, presence of other anomalies and development of fetal hydrops. We present four cases of congenital pulmonary airway malformation: antenatally detected by ultrasound with their follow-up in the post-natal period. Three of them have normal uneventful post-natal periods. One child was normal for six months of age then developed frequent respiratory tract infections.
Keywords: CPAM, USG, cystic lung lesion, Types of CPAM, respiratory tract infection
| Corresponding Author | How to Cite this Article | To Browse |
|---|---|---|
| , Post Graduate Resident, Md Radiodiagnosis, Nil Ratan Sircar Medical College, Wbuhs, West Bengal, India. Email:  |
Sahu S, Khokan M, Guha Roy A, A Case Series Report on Congenital pulmonary airway malformation – antenatal diagnosis and post-natal follow up. Int J Med Res Rev. 2024;12(4):126-131. Available From https://ijmrr.medresearch.in/index.php/ijmrr/article/view/1485 |
 |
 ©
©