The Double Trouble: Caudal Duplication Syndrome-A rare case series with a review of literature
Datta D1, Saha S2, Dakshit D3, Maity A4*, Bansal R5, Basu N6
DOI:https://doi.org/10.17511/ijmrr.2024.i02.05
1 Debarpita Datta, Senior Resident, Department of Radiodiagnosis, Medical College and Hospital , Kolkata, West Bengal, India.
2 Sudipta Saha, Associate Professor, Department of Radiodiagnosis, Medical College and Hospital, Kolkata, West Bengal, India.
3 Debashis Dakshit, Professor, Department of Radiodiagnosis, Medical College and Hospital , Kolkata, West Bengal, India.
4* Arup Maity, Senior Resident, Department of Radiodiagnosis, Medical College and Hospital, Kolkata, West Bengal, India.
5 Ruchi Bansal, Assistant Professor, Department of Radiodiagnosis, Medical College and Hospital, Kolkata, West Bengal, India.
6 Nupur Basu, Associate Professor, Department of Radiodiagnosis, Medical College and Hospital, Kolkata, West Bengal, India.
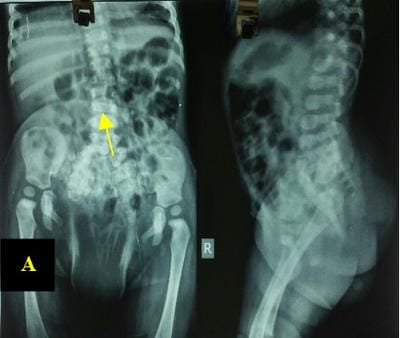
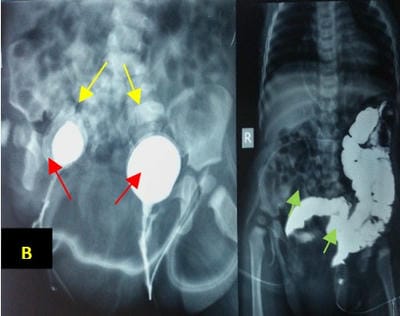
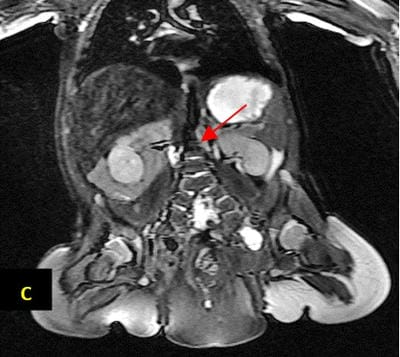
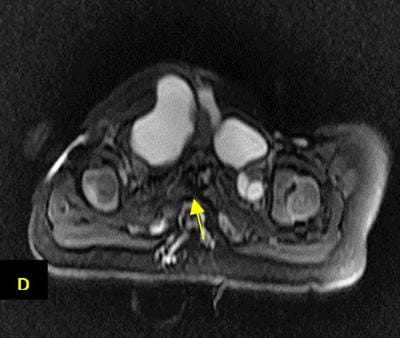
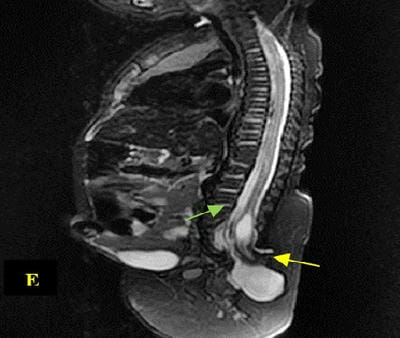
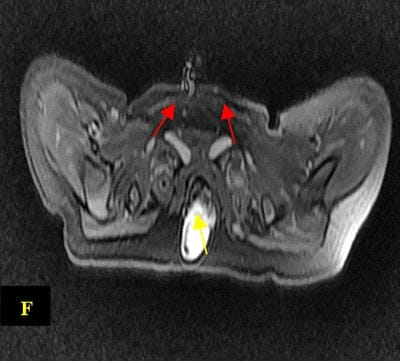
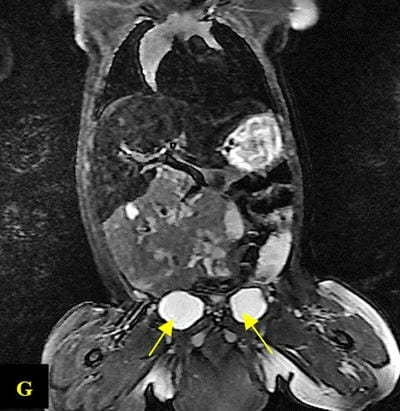
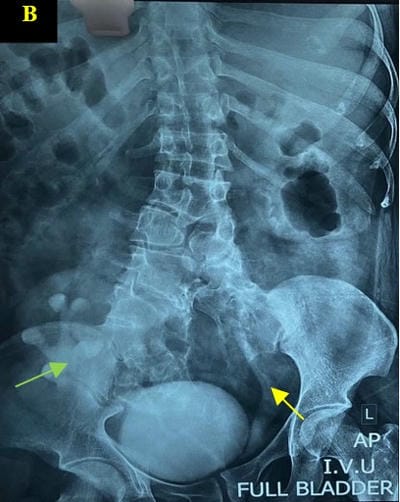
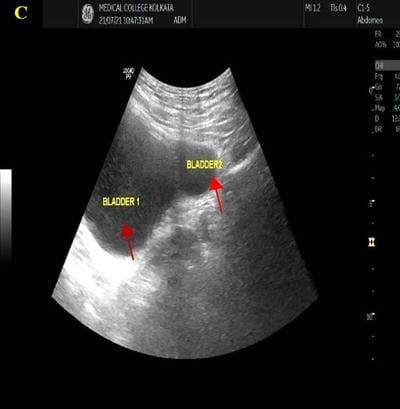
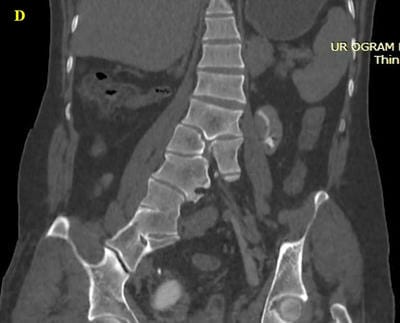
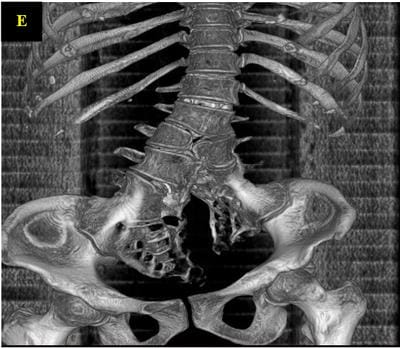
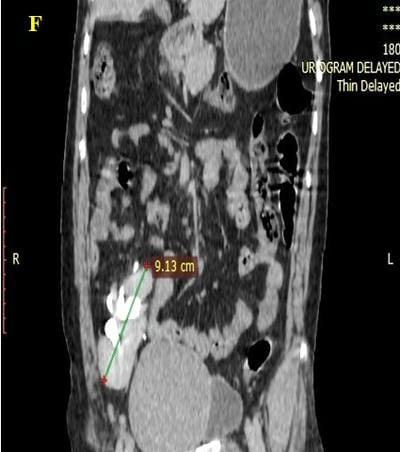
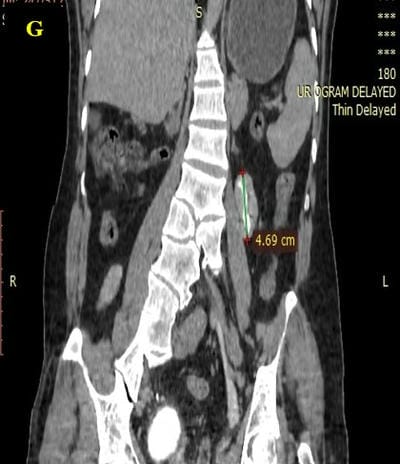
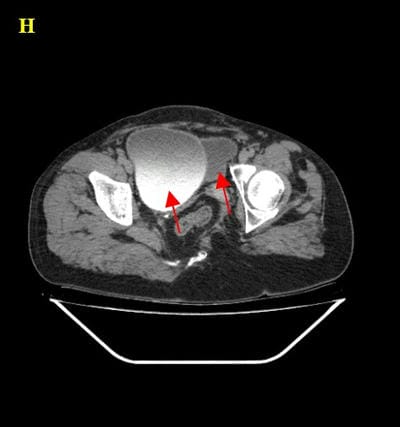
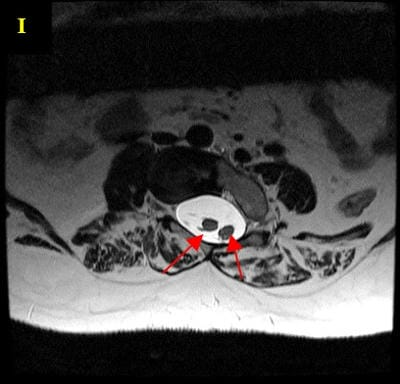
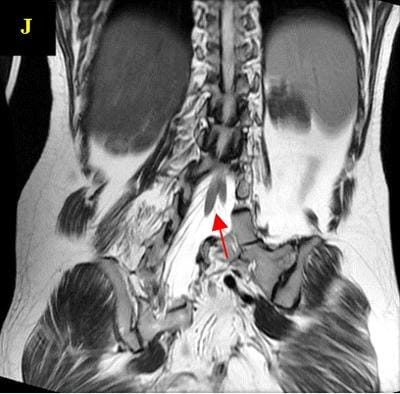
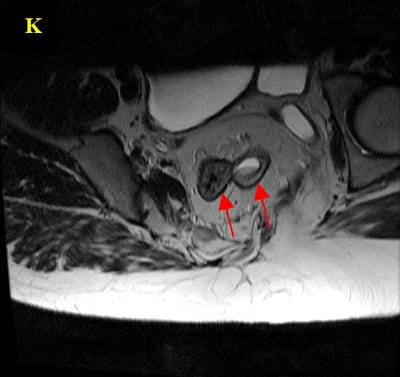
Caudal duplication syndrome (CDS) is a very rare disease entity with prevalence of less than 1 per 100,000 at birth. It includes a broad range of abnormalities and clinical symptoms, from single or partial duplication of the gastrointestinal, genitourinary and distal neural tube system organs to total duplication. There have been several ideas proposed to explain the complicated yet symmetrical abnormalities and the wide range of clinical manifestations of caudal duplication syndrome. In this case series, we present two confirmed cases of Caudal Duplication Syndrome in our institute. The management for this syndrome is individualized and may include surgical intervention to fuse or excise the duplicated organs.
Keywords: Caudal Duplication Syndrome, Duplication, Rare
| Corresponding Author | How to Cite this Article | To Browse |
|---|---|---|
| , Senior Resident, Department of Radiodiagnosis, Medical College and Hospital, Kolkata, West Bengal, India. Email:  |
Datta D, Saha S, Dakshit D, Maity A, Bansal R, Basu N, The Double Trouble: Caudal Duplication Syndrome-A rare case series with a review of literature. Int J Med Res Rev. 2024;12(2):57-64. Available From https://ijmrr.medresearch.in/index.php/ijmrr/article/view/1463 |
 |
 ©
©