True-Tales Of Ten T- prolymphocytic leukemia (T-PLL) Over A Decade
N Rajakumar N1*, H Vijay R2, Balikai G3, B Horatti L4, Shamnur M5
DOI: https://doi.org/10.17511/ijmrr.2023.i05.03
1* Namrata N N Rajakumar, Associate Professor, Pathology Molecular biology research, Kidwai Memorial Institute Of Oncology, Bangalore, Karnataka, India.
2 Raghavendra H Vijay, Professor, Department of Pathology, Kidwai Memorial Institute Of Oncology, Bangalore, Karnataka, India.
3 Girish Balikai, Professor, Department of Haematology, SDM College Of Medical Sciences And Hospital, Dharwad, Karnataka, India.
4 Laxmi B Horatti, Assistant Professor, Department Of Dermatology, Shri Atal Bihari Vajpayee Medical College And Research Center, Bangalore, Karnataka, India.
5 Murgesh Shamnur, Professor, Department Of Dermatology, JJM Medical College, Davangere, Karnataka, India.
Introduction: T-PLL is a mature T-cell leukemia typically presenting at stages of exponentially rising lymphocyte counts in peripheral blood, accompanied by splenomegaly and bone marrow involvement. They are rare and inherently aggressive and notoriously refractory to therapeutics. To our knowledge, this is the largest series of T-PLLs from India. Objectives; We studied Immunophenotypic characteristics, prognostic factors, outcomes, and treatments of 10 patients with T-PLL. Methods: Out of 4500 clinically suspected chronic leukemias, during 10 years, at Kidwai Memorial Institute of Oncology, which is a state cancer institute, diagnostic flow cytometric analysis was done and leukemias were classified based on WHO 2008 criteria, along with, morphology, cytogenetics, clinical, immunophenotyping and molecular findings. Results: out of 4500 cases of Chronic lymphoproliferative disorders sent for flow cytometric immunophenotyping, only 10 cases were diagnosed as T-PLL, accounting for 0.4 % mature leukemias of the lymphoid lineage. multiorgan involvement was common but effusion as a presenting feature was seen in only 10% of patients. Surprisingly skin involvement was evident in more number 70% of cases. single case showed cytogenetic abnormalities, later confirmed by FISH. Conclusions: Evaluation of the immunophenotype of this entity by flow cytometry is a critical part of diagnosis and is an indispensable tool in distinguishing T-PLL from other mature T-cell lymphoid neoplasms.
Keywords: T -prolymphocytic leukemia, immunophenotyping; T-leukemias. flow cytometry, Next generation sequencing
| Corresponding Author | How to Cite this Article | To Browse |
|---|---|---|
| , Associate Professor, Pathology Molecular biology research, Kidwai Memorial Institute Of Oncology, Bangalore, Karnataka, India. Email:  |
N Rajakumar N, H Vijay R, Balikai G, B Horatti L, Shamnur M, True-Tales Of Ten T- prolymphocytic leukemia (T-PLL) Over A Decade. Int J Med Res Rev. 2023;11(5):116-122. Available From https://ijmrr.medresearch.in/index.php/ijmrr/article/view/1432 |
 |
 ©
© 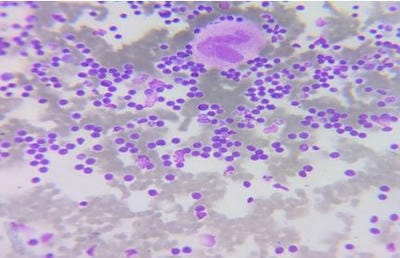 Figure 1: Bone marrow aspiration slides showing medium-sized prolymphocytes.
Figure 1: Bone marrow aspiration slides showing medium-sized prolymphocytes.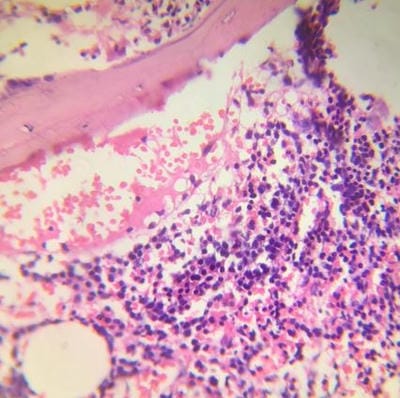 Figure 2: Bone marrow core biopsy revealing increase in prolymphocytes.
Figure 2: Bone marrow core biopsy revealing increase in prolymphocytes. Figure 3: Flow cytometric histogram showing Neoplastic cells( prolymphocytes) positive for CD markers of T cell type and negative for CD1a and Tdt.
Figure 3: Flow cytometric histogram showing Neoplastic cells( prolymphocytes) positive for CD markers of T cell type and negative for CD1a and Tdt.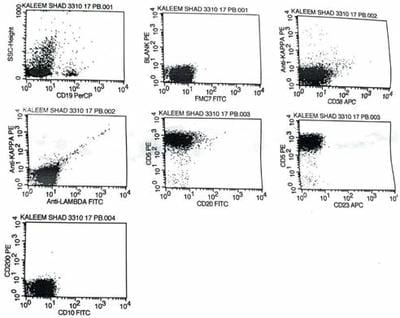 Figure 4: Flow cytometric histogram showing Neoplastic cells (Prolymphocytes)Negative for CD markers of B cell type.
Figure 4: Flow cytometric histogram showing Neoplastic cells (Prolymphocytes)Negative for CD markers of B cell type.